Foreword
This
publication contains recommendations from the Scientific Working Group for the
Analysis of Seized Drugs (SWGDRUG).
These recommendations are intended to assist forensic analysts and
managers in the development of analytical techniques, protocols and
policies. They are recognized to be
minimum standards that may be modified to address unique jurisdictional
requirements. SWGDRUG seeks to have these recommendations internationally
accepted as the foundation for good laboratory practice. These recommendations encompass Code of
Professional Practice, Education and Training, Methods of Analysis and Quality
Assurance. The SWGDRUG Core Committee
strongly urges the adoption of these recommendations by any laboratory involved
in the analysis of seized drugs.
Since 1997,
SWGDRUG has been working to provide useful and practical recommendations for
the analysis of seized drugs. SWGDRUG
recognizes that over time these recommendations may need to be updated as a
result of advances in technology, changes in accreditation requirements and/or
the emergence of new requirements. To
this end, SWGDRUG relies heavily on the input of the forensic community to
ensure that all recommendations remain useful and current. This synergetic approach is a key component
of the SWGDRUG process. I encourage
everyone to continue supporting the mission of SWGDRUG.
Finally, as
the Chair of SWGDRUG, I would be remiss if I did not single out several
individuals without whom SWGDRUG would not exist. Benjamin A. Perillo
conceived this working group and made it a reality. As former Chairs of SWGDRUG, Thomas J. Janovsky and Nelson A. Santos promoted and enhanced
SWGDRUG’s prominence in the Forensic Community.
Lastly, I recognize Sandra E. Rodriguez-Cruz, Secretariat, for her
untiring efforts in coordinating and facilitating the SWGDRUG meetings.
I would also
like to make special mention to the Office of National Drug Control Policy,
Counterdrug Technology Assessment Center and the National Institute of
Standards and Technology, which over the years have provided the financial
resources for SWGDRUG to operate.

Introduction
SWGDRUG
is comprised of a core committee of more than 20 forensic scientists from
around the world. The mission of SWGDRUG
is to recommend minimum standards for the forensic examination of seized drugs
and to seek their international acceptance.
SWGDRUG seeks to achieve this mission through the following objectives:
·
specifying
requirements for practitioners’ knowledge, skills and abilities,
·
promoting
professional development,
·
providing
a means of information exchange within the
forensic science community,
·
promoting
ethical standards of practitioners,
·
providing
minimum standards for examinations and reporting,
·
establishing
quality assurance requirements,
·
considering
relevant international standards, and
·
seeking international acceptance of SWGDRUG
recommendations.
Drug
abuse and trafficking in controlled substances are global problems, and
law enforcement has looked to international solutions
for these problems. In 1997 the U.S.
Drug Enforcement Administration (DEA) and the Office of National Drug Control
Policy (ONDCP) co-sponsored the formation of the Technical Working Group for
the Analysis of Seized Drugs (TWGDRUG).
Forensic scientists from the United States, England, Canada, Australia,
Japan, Germany and the Netherlands, as well as representatives of the United
Nations, several international forensic organizations and academia were invited
to meet in Washington, DC. This group, with input from around the world, developed educational
and professional development recommendations
for forensic practitioners. They
also developed quality assurance and identification recommendations for
seized drugs. The name Scientific Working Group for the
Analysis of Seized Drugs was adopted in 1999.
SWGDRUG
has received input from many forensic scientists in its recommendations development process. It has used
various methods of communication including its Internet site (www.swgdrug.org), presentations at
numerous local, national and international meetings, and personal
contacts. Following each meeting of the
Core Committee, updates are published and distributed.
SWGDRUG
sought and considered comments from the forensic science community on all its
proposals. In order for a recommendation
to be adopted, there are
specific procedures that must be met.
Please refer to the SWGDRUG’s bylaws, which can be found on the internet at www.swgdrug.org/bylaws.htm for additional details.
In July 2010
the leadership of SWGDRUG was transferred to Scott R. Oulton,
Chair and Sandra E. Rodriguez-Cruz, Secretariat. The various sub-committees continue to
research and develop proposals for additional recommendations with several
members completing their service to the group and
others replacing them by invitation.
The following chart details those persons who have
rendered service as members of the core committee over the years. For a list of current members, please
reference the SWGDRUG website.
Core
Committee
|
Ms. Susan Ballou
Office of Law Enforcement Standards
National Institute of Standards and Technology
Gaithersburg, Maryland
|
Dr. Yoshiteru Marumo
National Research
Institute of Police Science
Chiba, Japan
|
|
Dr. Suzanne Bell
West Virginia University
Morgantown, West Virginia
|
Mr. Jerry Massetti
California
Department of Justice California Criminalistics
Institute
Sacramento,
California
|
|
Mr. Robert Bianchi
Drug Enforcement Administration
McLean, Virginia
|
Mr. Christian Matchett
U.S. Army Criminal Investigation
Laboratory
Forest Park, Georgia
|
|
Mr. Joseph Bono (Secretariat)
Drug Enforcement Administration
Washington, DC
|
Dr. Iphigenia Naidis
United Nations Office on Drugs and Crime
Laboratory and Scientific Section
Vienna Austria
|
|
Dr. Michael
Bovens
Wissenschaftlicher
Dienst
Zurich,
Switzerland
|
Dr. Helmut
Neuman
Bundeskriminalamt
Forensic
Science Institute
Wiesbaden, Germany
|
|
Dr. Bob Bramley
Forensic Science Service
Birmingham, England
|
Mr. Osamu Ohtsuru
National Research Institute of Police Science
Chiba, Japan
|
|
Dr. Sylvia Burns
Forensic Science Service
Birmingham, England
|
Mr. Robert Ollis
Georgia Bureau of Investigation
Decatur, Georgia
|
|
Mr. Gary Chasteen
Los Angeles County Sheriff's Laboratory Scientific
Services Downey, California
|
Mr.
Scott R. Oulton (Secretariat and Chair)
Drug Enforcement Administration
Washington, DC
|
|
Mr. Alan B. Clark
Drug Enforcement Administration
Washington, DC
|
Mr. Richard A. Paulas
Illinois
State Police Forensic Sciences Command
Chicago,
Illinois
|
|
Mr. Jeffrey H. Comparin
(Secretariat)
Drug Enforcement Administration
Dulles, VA
|
Dr.
Eric Person
Department
of Chemistry
California
State University, Fresno
Fresno,
California
|
|
Dr. Alim A. Fatah
Office of Law Enforcement Standards
National Institute of Standards and Technology
Gaithersburg, Maryland
|
Mr. Benjamin Perillo
Drug Enforcement
Administration
Washington, DC
|
|
Dr. Maria Eugenia Forero Ruiz
National
Institute Legal Medicine and Forensic Science
Bogota, Colombia
|
Dr.
Karen Phinney
Analytical
Chemistry Division, NIST
Gaithersburg,
Maryland
|
|
Mr. Richard Gervasoni
Montgomery County Police Department Lab
Rockville, Maryland
|
Ms. Cate Quinn
Victoria
Police Forensic Services Department
Melbourne,
Australia
|
|
Ms. Jo Ann Given
Naval Criminal Investigative Service
Norfolk, Virginia
|
Ms. Pamela Reynolds
Federal Bureau of Investigation Laboratory
Quantico, Virginia
|
|
Mr. Garth Glassburg
Northeastern Illinois Regional Crime Laboratory
Vernon Hills, Illinois
|
Dr. Conrad Roberson
Georgia
Bureau of Investigation
Decatur,
Georgia
|
|
Ms. Dorothy Gordimer
Union County
Prosecutor’s Office Laboratory
Westfield, New
Jersey
|
Dr. Sandra E. Rodriguez-Cruz (Secretariat)
Drug Enforcement Administration
Vista, California
|
|
Ms.
Kathleen Higgins
Office
of Law Enforcement Standards
National
Institute of Standards and Technology
Gaithersburg,
Maryland
|
Mr. Nelson A. Santos (Chair)
Drug Enforcement Administration
Washington, DC
|
|
Dr.
Henk Huizer
Ministry
of Justice
Forensic
Science Laboratory
Rijswijk,
The Netherlands
|
Mr. Tshepo Shole
National Forensic Science Laboratory
Pretoria, South Africa
|
|
Dr.
Takako Inoue
National
Research Institute of Police Science
Tokyo,
Japan
|
Dr. Jay Siegel
Michigan State
University
School of Criminal
Justice
East Lansing,
Michigan
|
|
Ms. Linda Jackson
Virginia
Department of Forensic Science
Richmond, Virginia
|
Dr. Erkki Sippola
National Bureau of Investigation
Crime Laboratory
Vantaa, Finland
|
|
Mr.
Thomas J. Janovsky (Chair)
Drug
Enforcement Administration
Washington,
DC
|
Mr.
Zoran Skopec
Australian
Forensic Drug Laboratory
Pymble, NSW, Australia
|
|
Dr. Tohru Kishi
National Research Institute of Police Science
Chiba, Japan
|
Dr. Howard Stead
United Nations Office on Drugs
and Crime
Laboratory and Scientific Section
Vienna, Austria
|
|
Dr.
Cherif Kouidri
United
Nations International Drug Control Programme
Vienna,
Austria
|
Dr. Charles
"Chris" Tindall
Metropolitan State College of Denver Department of
Chemistry
Denver, Colorado
|
|
Mr. Richard Laing
Health
Canada
Drug
Analysis Service Laboratory
Vancouver,
Canada
|
Ms. Erin Trujillo
Los Angeles County
Sheriff’s Lab
Los Angeles,
California
|
|
Mr.
Marc LeBeau
Federal
Bureau of Investigation
Washington,
DC
|
Mr. Scott Vajdos
Harris County Institute of
Forensic Sciences
Houston, Texas
|
|
Mr.
John Lentini
Applied
Technical Services, Inc.
Marietta,
Georgia
|
Mr. Etienne van Zyl
National Forensic Science Laboratory
Pretoria, South Africa
|
|
Dr.
Bruce Lodge
Health
Canada
Ottawa,
Ontario Canada
|
Ms. Eileen Waninger
Federal
Bureau of Investigation Laboratory
Quantico, Virginia
|
|
Dr. Adriano Maldaner
Brazillian Federal Police
Brasilia, Federal District, Brazil
|
Mr. Jaco Westraat
National Forensic Science Laboratory
Pretoria, South Africa
|
|
Mr. Jack Mario
Suffolk
County Crime Laboratory
Hauppauge,
New York
|
Dr. Angeline Yap Tiong Whei
Health Sciences
Authority
Singapore
|
|
Dr. Yoshiteru Marumo
National Research
Institute of Police Science
Chiba, Japan
|
Dr. Udo Zerell
Bundeskriminalamt
Forensic Science
Institute
Wiesbaden, Germany
|
|
Dr.
Robert Powers
Connecticut
Department of Public Safety
Hartford,
Connecticut
|
|
PART I
A CODE OF PROFESSIONAL
PRACTICE FOR DRUG ANALYSTS
PREFACE
This Code of Professional Practice has been written specifically for
analysts. However, it is important that their managers and the
technicians and others who assist them in their work are equally aware of its
provisions, and they support the analyst in adhering to these. Where
appropriate, the provisions are also equally applicable to the technicians in
the approach to their own work.
1
Introduction
1.1
A Code of Professional Practice is intended to provide the
framework of ethical values and scientific and legal obligations within which
the analyst should operate. Details are also usually provided on how
alleged breaches of the Code will be investigated, what sanctions are available
and how appeals should be pursued.
1.2
A Code of Professional Practice is essential to analysts and their
managers in helping them carry out their duties in a proper manner and in
making appropriate decisions when questions of ethics arise.
1.3
A Code of Professional Practice that is enforced and publicly
available is also a powerful means of demonstrating the professional
expectations of analysts and the reliability of their findings to others in the
criminal justice system and the public at large.
1.4
SWGDRUG recommends that all employers of analysts develop a Code
of Professional Practice and the means of dealing with breaches of the
Code.
1.5
SWGDRUG further recommends that all Codes of Professional Practice
for analysts should include, as a minimum, provisions relating to their
professional conduct, their casework and the reporting of their results, as
provided in Section 2. For further information, see Supplemental Document SD-1 (Examples for Part I - A Code of
Professional Practice for Drug Analysts).
2
Code of professional practice
2.1 Professional
conduct
Analysts shall:
a) act with honesty, integrity and
objectivity;
b) work only within the bounds of their
professional competence;
c) take reasonable steps to maintain
their competence;
d) recognize that their overriding
duty is to criminal justice;
e) declare to their employer any prior
contact or personal involvement, which may give rise to conflict of
interest, real or perceived;
f) declare to their employer or other
appropriate authority any pressure intended to influence the result of an
examination.
2.2 Casework
Analysts shall:
a) strive to
demonstrate that the integrity and security of evidential materials and the
information derived from their analysis have been maintained while in their
possession;
b) strive
to have a clear understanding of what the customer
needs and all the necessary information, relevant evidential materials and
facilities available to reach a meaningful conclusion in an appropriate
timeframe;
c) employ an appropriate analytical
approach, using the facilities available;
d) make and retain full,
contemporaneous, clear and accurate records of all examinations and tests
conducted, and conclusions drawn, in sufficient detail to allow meaningful
review and assessment of the conclusions by an independent person competent in
the field;
e) accept responsibility for all
casework done by themselves and under their direction;
f) conduct all professional
activities in a way that protects the health and safety of themselves,
co-workers, the public and the environment.
2.3 Reporting
Analysts shall:
a) present advice and testimony,
whether written or oral, in an objective manner;
b) be prepared to reconsider and, if necessary, change their
conclusions, advice or testimony in light of new information or developments,
and take the initiative in informing their employer and customers promptly of
any such changes that need to be made;
c) take appropriate action if there is potential for, or there
has been, a miscarriage of justice due to new circumstances that have come to
light, incompetent practice or malpractice;
d) preserve customer confidentiality
unless officially authorized to do otherwise.
PART II
EDUCATION AND TRAINING
1 Introduction
Part
II recommends minimum education, training and experience for analysts
practicing in laboratories that conduct seized drug analyses. It describes the types of activities
necessary to continue professional development and reference literature
required in laboratories where they practice.
1.1
Recommendations listed in Part II are intended to apply
to any analyst who:
a) independently has access to unsealed evidential material in
order to remove samples for examination;
b) examines and analyzes seized drugs or related materials, or
directs such examinations to be done; and
c) as a
consequence of such examinations, signs reports for court or investigative
purposes.
2 Education
and experience for analysts
All new analysts shall have at least a bachelor’s
degree or equivalent (generally, a three to four year post-secondary degree) in
a natural/physical science. Coursework
shall include lecture and associated laboratory classes in general, organic and
analytical chemistry.
3 Continuing professional development
All
forensic scientists have an ongoing responsibility to remain current in their
field. In addition, laboratories shall
provide support and opportunities for continuing professional development. Minimum continuing professional development
requirements for a laboratory analyst are:
3.1 Twenty hours
of training every year.
3.2 Training
shall be relevant to the laboratory's mission.
Professional development may include training related to ancillary duty
assignments and supervision/management responsibilities.
3.3 Training
shall be documented.
3.4 Training can
be face-to-face interaction with an instructor, distance learning, self-directed
or computer based. Training can be provided from a variety of sources,
including, but not limited to the following:
·
chemistry
or instrumental courses taught at the post-secondary educational level
·
instrument
operation or maintenance courses taught by vendors
·
in-service
classes conducted by the employer
·
current
literature review
·
in-service
training taught by external providers
·
participation in relevant
scientific meetings or conferences (e.g., delivering an oral or poster
presentation, attending a workshop, providing reports on conferences).
4 Initial training requirements
These
minimum requirements allow individual laboratories to structure their training
program to meet their needs as it relates to type of casework encountered,
analytical techniques, available instrumentation and level of preparedness of
trainees.
4.1 There shall
be a documented training program, approved by laboratory management that
focuses on the development of theoretical and practical knowledge, skills and
abilities necessary to examine seized drug samples and related materials. The training program shall include the
following:
a) documented
standards of performance and a plan for assessing theoretical and practical
competency against these standards (e.g., written and oral examinations,
critical reviews, analysis of unknown samples and mock casework per topic
area);
b) a training syllabus providing
descriptions of the required knowledge and skills in specific topic areas in
which the analyst is to be trained, milestones of achievement, and methods of
testing or evaluating competency;
c) a period of
supervised casework representative of the type the analyst will be required to
perform;
d) a verification
document demonstrating that the analyst has achieved the required
competence.
4.2 Topic areas
in the training program shall include, as a minimum, the following:
·
relevant
background information on drugs of abuse (e.g., status of control and chemical
and physical characteristics)
·
techniques,
methodologies and instrumentation utilized in the examination of seized drug
samples and related materials
·
quality
assurance
·
ethics
·
expert/court
testimony and legal requirements
·
laboratory policy and
procedures (e.g., sampling, uncertainty, evidence handling, safety and
security) as they relate to the examination of seized drug samples and related
materials.
4.3 An individual
qualified to provide instruction shall have demonstrated competence in the
subject area and in the delivery of training.
5 References
and documents
The following references and documents shall
be available and accessible to analysts.
a) college/university level textbooks for
reference to theory and practice in key subject areas, e.g., general chemistry,
organic chemistry and analytical chemistry
b) reference
literature containing physical, chemical and analytical data. Such references include the Merck Index, Clarke’s Analysis of Drugs and Poisons, laboratory manuals of the
United Nations Drug Control Program, in-house produced spectra and published
standard spectra, (e.g., Mills and Roberson’s Instrumental Data For Drug Analysis, or
compendia from Pfleger or Wiley)
c) operation and maintenance manuals for each analytical
instrument
d) relevant periodicals (e.g., Journal of
Forensic Sciences, Forensic Science
International, Microgram, Journal of Canadian Society of Forensic Science, Japanese Journal of Forensic Science
and Technology)
e) laboratory
quality manual, standard operating procedures, and method validation and
verification documents
f) relevant
jurisdictional legislation (e.g., statutes and case law relating to controlled
substances, and health and safety legislation)
PART III A
METHODS OF
ANALYSIS/SAMPLING SEIZED DRUGS
FOR QUALITATIVE ANALYSIS
1 Introduction
This document
addresses minimum recommendations for sampling of seized drugs for qualitative
analysis.
NOTE For the purpose of this document the use of
the term “statistical” refers to “probability-based.”
1.1 The
principal purpose of sampling in the context of this recommendation is to
answer relevant questions about a population by examination of a portion of the
population (e.g., What is the net weight of the
population? What portion of the units of a population can be said to contain a
given drug at a given level of confidence?)
1.2 By
developing a sampling strategy and implementing appropriate sampling schemes,
as illustrated in Figure 1, a laboratory will minimize the total number of
required analytical determinations, while assuring that all relevant legal and
scientific requirements are met.

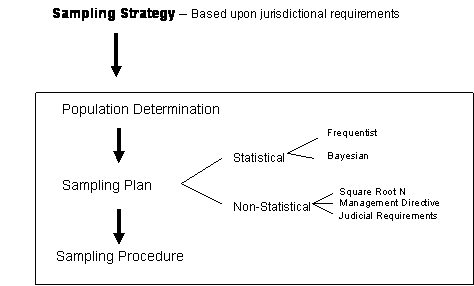
Figure 1: Relationship of the Various Levels Required in Sampling
2 Sampling strategy
An
appropriate sampling strategy is highly dependent on the purpose of the
investigation, the customer’s request, and the anticipated use of the results. Laws and legal practices form the foundation
of most strategies and shall be taken into account when designing a sampling
scheme. Therefore, specific sampling
strategies are not defined in this document.
2.1 The
laboratory has the responsibility to develop its own strategies consistent with
these recommendations. SWGDRUG
recommends attention to the following key points:
2.1.1 Sampling
may be statistical or non-statistical.
2.1.1.1 In
many cases, a non-statistical approach may suffice. The sampling plan shall provide an adequate
basis for answering questions of applicable law (e.g., Is
there a drug present in the population? Are statutory enhancement levels
satisfied by the analysis of a specified number of units?)
2.1.1.2 If
an inference about the whole population is to be drawn from a sample, then the
plan shall be statistically based and limits of the inference shall be
documented.
2.1.2 Statistically
selected units shall be analyzed to meet the SWGDRUG minimum recommendations
(see Part III B) for forensic drug identification if
statistical inferences are to be made about the whole population.
3 Sampling scheme
The sampling
scheme is an overall approach which includes population determination,
selection of the sampling plan and procedure and, when appropriate, sample
reduction prior to analysis (Figure 2).
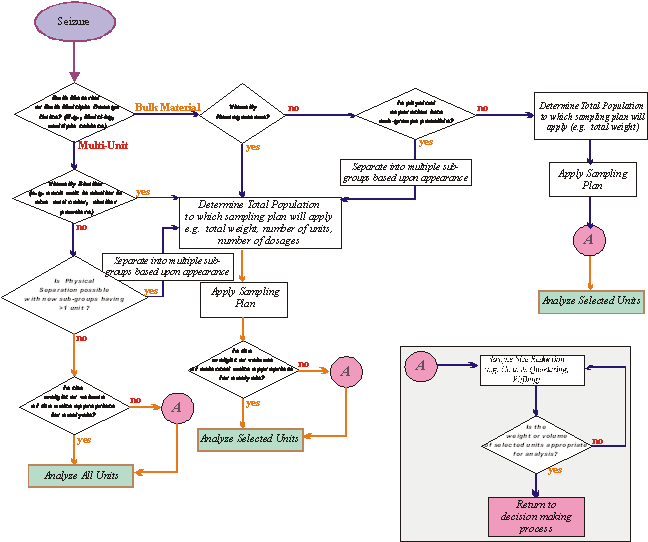 Insert A
Insert A
Figure 2: Example of a Sampling Scheme - A Decision
Flowchart
3.1 Population determination
3.1.1 The
population determination shall take into account all typical forms and
quantities in which exhibits may appear.
3.1.2 A
population can consist of a single unit or multiple units.
3.1.3 A
multiple unit population shall consist of items, which are similar in relevant
visual characteristics.
3.2 Sampling plan
There
are numerous sampling plans used in the forensic analysis of drugs, which are
applicable to single and multiple unit populations.
3.2.1 When
a single unit or bulk population is to be analyzed the issue of homogeneity
shall be addressed within the sampling plan.
3.2.1.1 One
sample is sufficient if the bulk material is homogeneous, or if it is made so
by the analyst.
3.2.1.2 If
the bulk material is not homogeneous, several samples from different locations
may be necessary to ensure that the test results are representative of the bulk
material and to avoid false negative results.
3.2.2 Depending
upon the inference to be drawn from the analysis for a multiple unit
population, the sampling plan may be statistical or non-statistical.
3.2.2.1 Statistical
approaches are applicable when inferences are made about the whole
population. For example:
a) The
probability that a given percentage of the population contains the drug of
interest or is positive for a given characteristic.
b) The
total net weight of the population is to be extrapolated from the weight of a
sample.
Published examples are provided below:
·
Frequentist
o
Hypergeometric
§
Frank
et al., Journal of Forensic Sciences, 1991, 36(2) 350-357
§
Guidelines on Representative Drug Sampling, European Network of Forensic Science
Institutes (ENFSI), 2009, www.enfsi.eu
§
American
Society for Testing and Materials (ASTM) E-2334
o
Other
probability based approaches
§
ASTM E105 “Standard Practice for Probability
Sampling of Materials”
§
ASTM E122 “Standard Practice for Calculating
Sample Size to Estimate, With a Specified Tolerable Error, the Average for a
Characteristic of a Lot or Process”
§
Guidelines on Representative Drug
Sampling,
ENFSI, 2009, www.enfsi.eu
·
Bayesian
o
Coulson et
al., Journal of Forensic Sciences, 2001, 46(6) 1456-1461
o
Guidelines on Representative Drug
Sampling,
ENFSI, 2009, www.enfsi.eu
3.2.2.2 Non-statistical
approaches are appropriate if no inference is to be made about the whole
population.
Examples
are provided below:
·
The
“square root” method
o
Recommended Methods for Testing Opium,
Morphine and Heroin: Manual for Use by National Drug Testing Laboratories, United Nations Office on Drugs and
Crime, 1998
·
Guidelines on Representative Drug
Sampling, ENFSI,
2009, www.enfsi.eu
·
Selection
of a single unit from a multiple unit population. This may be appropriate under certain
circumstances (e.g., management directives, legislative and/or judicial
requirements).
3.3 Sampling procedure
3.3.1 Establish
the procedure for selecting the number of units that will comprise the sample.
3.3.1.1 For
non-statistical approaches select a sample appropriate for the analytical
objectives.
3.3.1.2 For
statistical approaches SWGDRUG recommends that a random sampling be conducted.
3.3.2 Select
a random sample.
3.3.2.1 A
random sample is one selected without bias.
Computer generated random numbers or random number tables are commonly
employed for such tasks and these should be included in the sampling plan.
3.3.2.2 Random
sampling of items using random number tables may not be practical in all
cases. In these instances, an alternate
sampling plan shall be designed and documented to approach random
selection. A practical solution involves
a “black box” method, which refers to one that will prevent the sampler from consciously
selecting a specific item from the population (e.g., all units are placed in a
box and the samples for testing are selected without bias). Random sampling is discussed in the following
references:
·
ASTM E105
“Standard Practice for Probability Sampling of Materials”
·
Guidelines on Representative Drug Sampling, ENFSI, 2009, “Chapter 4:
Arbitrary Sampling ”, pages 9-10; www.enfsi.eu
3.4 Sample reduction
Sample
reduction may be applied in cases where the weight or volume of the selected
units is too large for laboratory analysis (Figure 2, insert A).
4 Analysis
4.1 Statistically selected sample(s)
SWGDRUG
recommends that each unit comprising the sample shall be analyzed to meet the
SWGDRUG minimum recommendations (Part III B) for
forensic drug identification, if statistical inferences are to be made about
the whole population.
4.2 Non-statistically selected sample(s)
SWGDRUG
minimum recommendations for forensic drug identification (Part III B) shall be applied to at least one
unit of the sample.
5 Documentation
Inferences
drawn from the application of the sampling plan and subsequent analyses shall
be documented.
6 Reporting
Sampling
information shall be included in reports (Part
IVA, Section 9.2).
6.1 Statistically selected sample(s)
Reporting
statistical inferences for a population is acceptable when testing is performed
on the statistically selected units as stated in Section 4.1 above. The language in the report must make it clear to the
reader that the results are based on a sampling plan.
6.2 Non-statistically selected sample(s)
The language in the report must make it clear
to the reader that the results apply to only the tested units. For example, 2 of 100 bags were analyzed and
found to contain Cocaine.
PART III B
METHODS OF ANALYSIS/DRUG
IDENTIFICATION
1 Introduction
The purpose
of PART III B is to recommend minimum standards for the forensic identification
of commonly seized drugs. It is
recognized that the correct identification of a drug or chemical depends on the
use of an analytical scheme based on validated methods (see PART
IV B – Validation)
and the competence of the analyst. It is
expected that, in the absence of unforeseen circumstances, an appropriate
analytical scheme effectively results in no uncertainty in reported identifications
(see PART
IV C – Uncertainty). SWGDRUG requires the use of multiple
uncorrelated techniques. It does not discourage the use of any
particular method within an analytical scheme and it is accepted that unique
requirements in different jurisdictions may dictate the practices followed by a
particular laboratory.
2 Categorizing analytical techniques
Techniques
for the analysis of drug samples are classified into three categories (see
Table 1) based on their maximum potential discriminating power. However, the classification of a technique
may be lower, if the sample, analyte or mode of
operation diminishes its discriminating power.
Examples of
diminished discriminating power may include:
- an infrared spectroscopy
technique applied to a mixture which produces a combined spectrum
- a mass spectrometry technique
which only produces molecular weight information
Table
1: Categories of Analytical Techniques
|
Category A
|
Category B
|
Category C
|
|
Infrared
Spectroscopy
|
Capillary
Electrophoresis
|
Color Tests
|
|
Mass
Spectrometry
|
Gas
Chromatography
|
Fluorescence
Spectroscopy
|
|
Nuclear
Magnetic Resonance Spectroscopy
|
Ion
Mobility Spectrometry
|
Immunoassay
|
|
Raman
Spectroscopy
|
Liquid
Chromatography
|
Melting
Point
|
|
X-ray Diffractometry
|
Microcrystalline
Tests
|
Ultraviolet
Spectroscopy
|
|
|
Pharmaceutical
Identifiers
|
|
|
|
Thin Layer
Chromatography
|
|
|
|
Cannabis
only:
Macroscopic Examination
Microscopic Examination
|
|
3 Identification criteria
SWGDRUG
recommends that laboratories adhere to the following minimum standards:
3.1 When
a validated Category A technique is incorporated into an analytical scheme, at
least one other technique (from either Category A, B or C) shall be used.
3.2
When
a Category A technique is not used, at least three different validated
techniques shall be employed. Two of the three techniques shall be based on
uncorrelated techniques from Category B.
3.2.1 For cannabis, macroscopic and
microscopic examinations will be considered as uncorrelated techniques from
Category B when observations include documented details of botanical
features. Laboratories shall define the
acceptance criteria for these features for each examination.
3.2.2 For exhibits of cannabis that lack
sufficient observable macroscopic and microscopic botanical detail (e.g.
extracts or residues), D9-tetrahydrocannabinol
(THC) or other cannabinoids shall be identified
utilizing the principles set forth in sections 3.1 and 3.2.
3.3
Botanists may identify cannabis and other
botanical material utilizing morphological characteristics (category B) alone provided sufficient botanical
features appropriate for identification are observed. Such examinations shall be made only by
analysts competent in botanical identifications. In this context botanical competence applies
to those examiners recognized as professional botanists or those assessed to be
competent by such. Identifications of chemical components contained in
botanicals (mescaline, opiates, psilocin, etc.) should rely on principles set
forth in sections 3.1 and 3.2.
3.4
All
Category A and botanical identifications shall have data that are reviewable.
Where a Category A technique is not used, the
requirement for reviewable data applies to category B techniques. Examples of reviewable data are
·
printed
spectra, chromatograms, digital images, photographs or photocopies (color,
where appropriate) of TLC plates
·
contemporaneous
documented peer review for microcrystalline tests
·
reference
to published data for pharmaceutical identifiers
·
For cannabis and botanical materials
only: recording of detailed descriptions of morphological characteristics.
3.5
For
the use of any method to be considered of value, test results shall be
considered “positive.” While “negative”
test results provide useful information for ruling out the presence of a
particular drug or drug class, these results have no value toward establishing
the forensic identification of a drug.
3.6
The
laboratory shall employ quality assurance measures to ensure the results
correspond to the exhibit. Example measures are:
·
the
use of two separate samplings
·
sample
identification procedures such as bar-coding and witness checks
·
good
laboratory practices (e.g., positive and negative controls, one sample opened
at a time, procedural blanks)
3.7
In
cases where hyphenated techniques are used (e.g. gas chromatography-mass
spectrometry, liquid chromatography-diode array ultraviolet spectroscopy), they
will be considered as separate techniques provided that the results from each
are used.
3.8
The
chosen analytical scheme shall demonstrate the identity of the specific drug
present and shall preclude a false positive identification and minimize false
negatives. Where a scheme has limitations, this shall be reflected in the final
interpretation (see Part IVC - Uncertainty).
4 Comment
These
recommendations are minimum standards for the forensic identification of
commonly seized drugs. However, it
should be recognized that they may not be sufficient for the identification of
all drugs in all circumstances. Within
these recommendations, it is up to the individual laboratory’s management to
determine which combination of analytical techniques best satisfies the
requirements of its jurisdiction.
PART III C
METHODS OF ANALYSIS/CLANDESTINE DRUG LABORATORY EVIDENCE
These recommendations are
intended to be used in conjunction with the general requirements for the
analysis of seized drugs. This document
provides guidance on the chemical analysis of items and samples related to
suspected clandestine drug laboratories.
It does not address scene attendance or scene processing. This document provides general recommendations
for the analysis of clandestine laboratory evidence and is not a substitute for
detailed and validated laboratory policies and technical procedures.
1
Introduction
1.1
SWGDRUG considers an understanding of clandestine laboratory
synthetic routes and the techniques used in the analysis of related samples to
be fundamental to the interpretation and reporting of results. This understanding assures that results and
conclusions from methods are reliable and analytical schemes are fit for
purpose.
1.2
The qualitative and quantitative analyses of clandestine laboratory
evidence can require different approaches relative to routine seized drug
analyses. Analysts shall understand the
limitations of the procedures used in their qualitative and quantitative analyses.
1.3
Laboratory management shall ensure that clandestine laboratory
synthesis and analysis training be provided through relevant procedures,
literature, and practical experience.
Practical experience typically includes production, sampling and analysis
of clandestine laboratory training samples.
1.4
Laboratory management shall ensure that chemical safety and hygiene
plans address and mitigate hazards associated with clandestine laboratory
evidence.
1.5
Laboratory management shall consider customer / local requirements
which influence the application of these recommendations.
2
Safety
2.1
Many items seized at clandestine laboratories may be intrinsically
dangerous. These may include items of
unknown composition and chemicals that have not been fully characterized and
whose specific hazards are not known.
Therefore, caution must be exercised and routine safety protocols may
not be sufficient.
2.2
The following are required in addition to the routine laboratory
safety program in place for the analysis of seized drugs (see Part IVA – Health and Safety):
·
safety procedures and in the use of safety and protective equipment
for all staff responsible for handling items
·
protective breathing equipment
·
listings of the relevant hazards (e.g. MSDS) associated with
components commonly found at clandestine laboratory sites and knowing what they
mean
·
accident prevention, emergency response procedures, and incident
reporting protocols
2.3
The handling, analysis, and storage of items seized from clandestine
laboratories require additional procedures, facilities and equipment. (see Part IVA – Physical Plant): Examples are:
·
specialized ventilation
equipment (e.g. fume hoods) to prevent exposure to harmful fumes and
vapors
·
provision of personal
protective equipment such as safety glasses, chemical resistant gloves,
laboratory coats, respirators, face masks, and air monitors
·
maintenance of a clean,
uncluttered workspace
·
specialized emergency
equipment stations
·
chemical disposal and
destruction facilities and procedures
·
specialized evidence receipt,
storage and disposal requirements designed to mitigate expected dangers (e.g.
limited sample size, proper packaging of reactive materials, use of absorbents,
properly ventilated storage)
2.4
Analysts shall be aware of the hazards associated with clandestine
laboratories samples. Examples are:
·
extracting from strong acids
and bases (e.g. hydriodic acid, sodium hydroxide)
·
handling fuming acids and
bases (e.g. hydrochloric acid, ammonia)
·
poisonous gasses (e.g. phosphine, chlorine, hydrogen sulfide) and their potential
release from evidence during analysis
·
poisonous, carcinogenic, and
mutagenic materials (e.g. mercuric chloride, chloroform, potassium cyanide)
·
reactive and air sensitive
materials (e.g. white phosphorus, lithium)
·
potential testing
incompatibilities (e.g. phosphorus with Raman, color test reagents with cyanide
salts, exothermic reactions)
·
radioactive materials (e.g.
thorium)
·
volatile and flammable
solvents (e.g. acetone, diethyl ether, methylated
spirits)
3
Sample selection for analysis
3.1
The primary purpose of analysis is to prove or disprove allegations
of clandestine drug syntheses.
Accordingly, analysts must select items which relate to the
manufacturing process.
3.2
Not all items seized at a clandestine laboratory site may need to be
analyzed. It is recommended that
information be shared between the analyst and on-scene personnel to aid in
sample selection.
3.3
Items should be selected for analysis, based on jurisdictional
requirements, and which are likely to contain:
·
finished product
·
intermediates
·
precursors
·
key reagents
·
reaction mixtures
3.4
Some of the following types of items may be analyzed as they can
assist in determining the chemical reaction(s) undertaken and the scope of the
clandestine laboratory:
·
materials that appear to be
waste
·
unlabeled materials that
appear to be contaminated solvents, acids, or bases
·
samples from contaminated
equipment
3.5
Items that are readily obtained from local retail stores and are
sold from reputable manufacturers/distributors may not need to be analyzed,
particularly if collected from sealed and labeled containers. These include:
·
solvents (e.g. toluene,
mineral spirits)
·
acids (e.g. hydrochloric
acid, sulfuric acid)
·
bases (e.g. sodium hydroxide,
ammonia water)
4
Analysis
4.1
Substances whose presence are reported or contribute to formulating
reported conclusions shall be identified with an adequate analytical scheme.
4.2
Where possible, the identification of organic compounds shall follow
the guidelines for the analysis of seized drugs (see Part III B).
4.3
The discriminating power of analytical techniques for the
identification of inorganic materials depends on the particular analyte. In each
case the analytical scheme shall:
·
have sufficient
discriminating power to identify the material to the exclusion of others (e.g.
identification of both the cation and anion in salts)
·
utilize two or more
techniques, preferably from different analytical groups described below
4.4
The following list of analytical groups and techniques are in no
particular order and are not exhaustive.
Analytical techniques must be selected which provide sufficient
discriminating power for each analyte. Some techniques may not be useful for
particular analytes and each must be evaluated to
determine suitability.
4.4.1
Analytical Group 1: Elemental Analysis Techniques – these techniques may
provide positive results for elements present in a sample but typically require
additional tests to distinguish forms (e.g. oxidation state).
·
X-Ray Fluorescence (XRF)
·
Energy Dispersive X-Ray Detectors
for Scanning Electron Microscopes (SEM-EDX)
·
Atomic Absorption
Spectroscopy
·
Atomic Emission Spectroscopy
and Flame Tests (an attached spectrometer significantly increases the
discriminating power relative to flame tests)
·
Mass Spectrometry (utilizing
Inductively Coupled Plasma sources or for elements with unique isotopic
abundance patterns)
4.4.2
Analytical Group 2: Structural Elucidation Techniques – these techniques may have
high discriminating power for polyatomic analytes.
·
Nuclear Magnetic Resonance
(NMR)
·
Infrared Spectroscopy (IR and
FTIR)
·
Raman Spectroscopy
·
UV-Vis & Fluorescence
Spectroscopy
·
Mass Spectrometry
4.4.3
Analytical Group 3: Separation Techniques – these techniques can be
valuable for mixtures and for distinguishing different forms of an element
(e.g. phosphate and phosphite).
·
Capillary Electrophoresis
·
Ion Chromatography
·
Liquid Chromatography
·
Gas Chromatography
·
Thin Layer Chromatography
4.4.4
Analytical Group 4: Chemical Properties – These techniques involve
observations of chemical changes.
Utilizing several of these techniques, in series or combination, can
often increase discriminating power.
·
Solubility and miscibility
tests
·
Spot and precipitation tests
·
Microcrystalline tests
·
pH (of liquids or vapors)
·
Radioactive decay
·
Flammability
·
Reactivity with water, air,
or other materials
4.4.5
Analytical Group 5: Physical Properties – These techniques involve
observations of physical properties. The
discriminating power of these techniques depends on the measuring device.
·
Physical state or states
·
Phase transitions including
melting points, boiling points, sublimation temperature and vapor pressure
·
Density (relative density and
density of mixtures have reduced discriminating power)
·
Color
·
Viscosity and surface tension
·
Refractive index
·
Crystal forms measured with
polarized light microscopy or x-ray diffraction techniques
4.5
If limited or qualified conclusions are sufficient (e.g. basic
aqueous layer, non-polar organic solvent, a material containing the element
phosphorus), tests of limited discriminating power may be utilized within an
analytical scheme.
4.6
Analytical reference materials may not be available for the analysis
of intermediates and byproducts. In
these cases, samples taken from a test reaction in conjunction with suitable
reference literature may be used for comparison purposes.
4.7
Quantitative measurements of clandestine laboratory samples have an
accuracy which is dependent on sampling and, if a liquid, on volume
calculations. Accordingly, these
measurements and calculations may be based on estimates. Under these conditions, a rigorous
calculation of measurement uncertainty is often not possible or necessary and
the uncertainty may best be conveyed by using a qualifier statement on the
report (e.g. approximately, not to exceed, no less than).
5
Yield and capacity calculations
5.1
Yield and capacity calculations can be achieved from a number of
approaches and shall be based on relevant case information, suitable
literature, laboratory and jurisdictional requirements.
5.2
Reported yields and capacities shall be based upon information
documented in the laboratory case file.
5.3
Calculated yields can be expressed as theoretical or expected.
5.3.1
SWGDRUG recommends that reported yields be accompanied with an
explanation clarifying the limitations or considerations.
5.3.1.1
Theoretical yields are calculated based on the amount of known
chemical, the stoichiometry of the reaction used in
the clandestine laboratory and the product.
Theoretical yields are not achievable in practice and their reporting
can be misinterpreted.
5.3.1.2
Expected yields are calculated based upon published data,
experience, or practical experimentation.
Expected yields can be highly variable based upon the factors listed
below.
5.4
In calculating expected yields and capacities in clandestine
laboratories, many different sources of information can be used. Each case is different and will have a
different set of evidence from which to draw information, including, but not
limited to:
·
amounts of finished products,
precursors, or essential chemicals present
·
amount of waste present
·
size of reaction vessels and
equipment
·
volume and quantity of
containers
·
type / quantity of equipment
and chemicals used
·
state of equipment and
premises (e.g. cleanliness of site and equipment)
·
the apparent skill and
laboratory practice of the operator
·
the procedures (i.e. recipe)
followed by the operator
5.5
In addition to observations about the clandestine laboratory site
itself, other pieces of evidence can lead to an understanding of yields and
capacities, including, but not limited to:
·
length of time the laboratory
has been in operation
·
intercepted conversations
·
statements made by the
clandestine laboratory operator during an interview/interrogation
·
documents describing
purchases of equipment, precursors, or reagents
·
photographs of the clandestine laboratory site and other related areas.
·
records kept by the
clandestine laboratory operator (e.g. seized recipes or records of previously
manufactured quantities)
5.6
When calculating capacity, ensure that the values were not obtained
from the same source (e.g. empty blister packs and tablet waste).
6
Reports and conclusions
6.1
Communications and reports, either written or verbal, shall be based
upon all of the available and relevant information and with clearly stated
assumptions and conditions.
6.2
There are many facets to a clandestine laboratory investigation,
such as:
·
the illicit drug being made
·
the synthetic route being
utilized
·
the type of equipment found
at the site
·
the past/potential production
at the site
·
the final form of the illicit
drug
·
the batch size at the site
·
whether a tabletting
/ encapsulating operation was present
6.3
Factors to consider in determining what to report include, but are
not limited to:
·
jurisdictional requirements
·
governing body (agency)
requirements
·
customer requests
·
potential exculpatory
information
·
samples / analytes
which represent the multiple stages in a reaction process
6.4
Laboratories should have documented policies establishing protocols
for reviewing verbal information and conclusions should be subject to technical
review whenever possible. It is
acknowledged that responding to queries in court or investigative needs may
present an exception.
6.5
When technical reviews are conducted, the individual reviewing the
conclusions must be knowledgeable in the processing, analysis, and reporting of
clandestine laboratory seizures.
7
Training
7.1
Analysis and interpretation of a clandestine laboratory case
requires specialized skills. The main
objective of clandestine laboratory training programs should be to provide new
analysts with a sound education in the fundamental areas of clandestine
laboratory evidence analysis. These
guidelines assume the student is qualified as a seized drug analyst.
7.2
Analysts shall receive training which will enable them to safely
perform the analysis of clandestine drug laboratory samples.
7.3
Analysts shall receive training which will enable them to assist in
investigation of clandestine drug syntheses.
Aspects of this training may include:
·
chemical separation
techniques (e.g. acid/base extractions, ion pair extractions, precipitation)
·
production estimates
·
study of pertinent drug
syntheses by various routes
·
training on intermediates and
route specific by-products
·
knowledge of common and alternative
sources of chemicals
·
training in inorganic
chemistry, analysis techniques, and interpretation
·
common terminology used in
organic chemistry and synthesis
·
application of critical
thinking and problem solving skills to the evaluation of all case information
(e.g. officer and scene reports, recipes, chemical data)
·
the ability to recognize when
additional information is required, identify sources for that information
(journals, monographs, underground references), critically evaluate the
reference and apply that knowledge to case information
·
legal issues and courtroom
testimony
7.4
Analysts should stay current in the field of clandestine drug
manufacturing and clandestine laboratory investigations. Examples of this element include:
·
joining regional, national,
and international scientific organizations
·
attending conferences
specializing in clandestine drug manufacture
·
receiving training by
qualified instructors covering current trends and reviews
·
reading pertinent scientific
literature
·
monitoring relevant illicit
literature and sites
PART IV A
QUALITY ASSURANCE/GENERAL PRACTICES
It is the goal of a laboratory's drug analysis program to provide the customers
of the laboratory's services access to quality drug analysis. It is the goal of these recommendations in
PART IV A to provide a quality framework for management of the processing of
drug casework, including handling of evidentiary material, management
practices, analysis and reporting. These
are minimum recommendations for practice.
The term
“evidence” has many meanings throughout the international community. In this document it is used to describe drug
exhibits that enter a laboratory system.
2 Quality management system
A documented quality
management system shall be established and maintained.
2.1 Personnel
responsible for this shall be clearly designated and shall have direct access
to the highest level of management concerning laboratory policy.
2.2
The
quality management system shall cover all procedures and reports associated
with drug analysis.
3 Personnel
3.1 Job description
The
Job descriptions for all personnel should include responsibilities, duties and
required skills.
3.2 Designated personnel and responsibilities
An
individual (however titled) may be responsible for one or more of the following
duties:
3.2.1
Quality Assurance Manager: A designated person who is
responsible for maintaining the quality management system (including an annual
review of the program) and who monitors compliance with the program.
3.2.2
Health & Safety Manager: A designated person who is
responsible for maintaining the Laboratory Health and Safety program (including
an annual review of the program) and monitors compliance with the program.
3.2.3
Technical Support Personnel: Individuals who perform
basic laboratory duties, but do not analyze evidence.
3.2.4
Technician/Assistant Analyst: A person who analyzes
evidence, but does not issue reports for court purposes.
3.2.5
Analyst: A designated person who:
a) examines and
analyzes seized drugs or related materials, or directs such examinations to be
done
b) independently has access to unsealed evidence in order to
remove samples from the evidentiary material for examination AND
c) as a consequence of such examinations, signs reports for
court or other purposes.
3.2.6
Supervisor: A designated person who has the overall
responsibility and authority for the technical operations of the drug analysis
section. Technical operations include,
but are not limited to protocols, analytical methodology, and technical review
of reports.
3.3
Qualifications/Education
3.3.1
Technical
Support Personnel shall
a) have education, skills and abilities commensurate with their
responsibilities AND
b) have on-the-job training specific to their position.
3.3.2
Technicians/Assistant
Analysts shall
a) have education, skills and abilities commensurate with their
responsibilities AND
b) have on-the-job training specific to their position.
3.3.3
Analysts
shall meet educational requirements stated in PART
II – Education and Training (Section 2).
3.3.4
Supervisors
shall
a) meet all the requirements of an analyst (3.3.3),
b) have a minimum of two (2) years of experience as an analyst
in the forensic analysis of drugs and
c) demonstrate knowledge necessary to evaluate analytical
results and conclusions.
3.4
Initial training requirements
Initial training requirements for
analysts are defined in PART II – Education and Training
(Section 4).
3.5 Maintaining competence
Continuing
professional development for analysts is defined in PART
II – Education and Training (Section 3).
4 Physical plant
4.1
Laboratories
shall provide a healthy, safe and secure environment for its personnel and
operations.
4.2
Laboratories
shall contain adequate space to perform required analytical functions and
prevent contamination.
4.3
Chemical
fume hoods shall be provided. They shall
be properly maintained and monitored according to an established schedule.
4.4
A
laboratory cleaning schedule should be established and implemented.
4.5
Adequate
facilities shall be provided to ensure the proper safekeeping of evidence,
standards and records.
4.6
Appropriately
secured storage shall be provided to prevent contamination of chemicals and
reagents.
5 Evidence control
Laboratories
shall have and follow a documented evidence control system to ensure the
integrity of physical evidence.
5.1
Receiving and identifying evidence
Laboratories shall maintain records of
requests for analysis and of the respective items of evidence. A unique
identifier shall be assigned to each case file or record. For chain-of-custody purposes, the evidence
shall be compared to the submission documentation, any significant observations
of irregularity shall be documented in the case file or record, and the
submitter informed promptly. This file
or record shall include, at least, the following:
·
submission
documents or copies
·
identity
of party requesting analysis and the date of request
·
description
of items of evidence submitted for analysis
·
identity
of the person who delivers the evidence, along with date of submission
·
for
evidence not delivered in person, descriptive information regarding mode of delivery
and tracking information
·
chain
of custody record
·
unique case identifier.
5.2 Integrity
of evidence
Evidence shall be properly secured
(e.g., sealed). Appropriate storage
conditions shall ensure that, insofar as possible, the composition of the
seized material is not altered. All
items shall be safeguarded against loss or contamination. Any alteration of the evidence (e.g.
repackaging) shall be documented.
Procedures shall be implemented to assure that samples are and remain
properly labeled throughout the analytical process.
5.3
Storage of evidence
Access to the evidence storage area
shall be granted only to persons with authorization and access shall be
controlled. A system shall be
established to document a chain of custody for evidence in the laboratory.
5.4 Disposition
of evidence
Records shall be kept regarding the
disposition (e.g., return, destruction, conversion to another use) of all items
of evidence.
5.5 Documentation
retention procedures
All laboratory records such as
analytical results, measurements, notes, calibrations, chromatograms, spectra
and reports shall be retained in a secure fashion in accordance with
jurisdictional requirements.
6 Analytical
procedures
6.1
Analytical procedures for drug
analysis
6.1.1
Laboratories
shall have and follow documented analytical procedures.
6.1.2
Laboratories
shall have in place protocols for the sampling of evidence (See PART
III A – Sampling).
6.1.3
Work
practices shall be established to prevent contamination of evidence during
analysis.
6.1.4
Laboratories
shall monitor the analytical processes using appropriate controls and traceable
standards.
6.1.5
Laboratories
shall have and follow documented guidelines for the acceptance and
interpretation of data.
6.1.6
Analytical
procedures shall be validated in compliance with PART
IV B - Validation.
6.1.7
When
analysts determine the identity of a drug in a sample, they shall employ
quality assurance measures to ensure the results correspond to the
exhibit. (See Part III B – Drug Identification)
6.2
Verification of drug reference
materials
6.2.1
The
identity of certified reference materials shall be verified prior to their
first use.
6.2.2
The
identity of uncertified reference materials shall be authenticated prior to use
by methods such as mixed melting point determination, Mass Spectrometry,
Infrared Spectroscopy, or Nuclear Magnetic Resonance Spectroscopy.
6.2.3
Verification
shall be performed on each new lot of drug reference material.
6.2.4
All
verification testing shall be documented.
The documentation shall include the name of the individual who performed
the verification, date of verification, verification test data and
reference used in verification.
7 Instrument/Equipment performance
7.1
Instrument performance
Instruments shall be routinely
monitored to ensure that proper performance is maintained.
7.1.1
Monitoring
should include the use of reference materials, test mixtures, calibration
standards, blanks, etc.
7.1.2
Instrument
performance monitoring shall be documented.
7.1.3
The
manufacturer's operation manual and other relevant documentation for
instrumentation should be readily available.
7.2
Equipment
7.2.1
Only
suitable and properly operating equipment shall be employed.
7.2.2
Equipment
performance parameters should be routinely monitored and documented.
7.2.3
The
manufacturer's operation manual and other relevant documentation for each piece
of equipment should be readily available.
8 Chemicals
and reagents
8.1
Chemicals
and reagents used in drug testing shall be of appropriate grade for the tests
performed.
8.2
There
shall be documented formulations for all chemical reagents produced within the
laboratory.
8.3
Documentation
for reagents prepared within the laboratory shall include identity,
concentration (when appropriate), date of preparation, identity of the
individual preparing the reagents, storage conditions (if appropriate) and the
expiration date (if appropriate).
8.4
The
efficacy of all test reagents shall be checked prior to their use in casework.
Results of these tests shall be documented.
8.5
Chemical
and reagent containers should be dated and initialed when received and also
when first opened.
8.6
Chemical
and reagent containers shall be labeled as to their contents.
9 Casework documentation, report writing and review
9.1
Casework documentation
9.1.1
Documentation
shall contain sufficient information to allow a peer to evaluate case notes and
interpret the data.
9.1.2
Evidence
handling documentation shall include chain of custody, information regarding
packaging of the evidence upon receipt, the initial weight/count of evidence to
be examined (upon opening), a description of the evidence and communications
regarding the case.
9.1.3
Analytical
documentation should include procedures, standards, blanks, observations, test
results and supporting documentation including charts, graphs and spectra
generated during an analysis.
9.1.4
Casework
documentation shall be preserved according to documented laboratory policy.
9.2
Report
writing
Reports issued by laboratories shall be accurate,
clear, objective, and meet the requirements of the jurisdictions served.
These reports shall include the following
information:
• title of report
• identity and location of the testing laboratory
• unique case identifier (on each page)
• clear identification of the end of the report (e.g., Page 3
of 3)
• submitting
agency
• date of receipt of evidence
• date of report
• descriptive list of submitted evidence
• identity and signature (or electronic equivalent) of analyst
• results / conclusions
• a list of analytical techniques employed
• sampling (See Part IIIA, Section 6 Reporting)
• uncertainty (see Part IVC
Uncertainty).
If elements listed above are not included on the
report, the laboratory shall have documented reasons (i.e. specific
accreditation, customer or jurisdictional considerations), for not doing so.
9.3
Case review
9.3.1
Laboratories
shall have documented policies establishing protocols for technical and
administrative case review.
9.3.2
Laboratories
shall have a documented policy for resolving case review disagreements between
analysts and reviewers.
10 Proficiency and competency
testing
Each
laboratory shall establish a documented competency testing and proficiency
testing program. Each laboratory shall
have documented protocols for monitoring the competency and proficiency of its
analysts.
NOTE It is recognized that different jurisdictions
may define competency and proficiency testing in a manner other than how they
are used here. In this context,
competency tests measure the ability of the analyst to produce accurate
results. Proficiency tests are an
ongoing process in which a series of proficiency samples, the characteristics
of which are not known to the participants, are sent to laboratories on a
regular basis. Each laboratory is tested
for its accuracy in identifying the presence (or concentration) of the drug
using its usual procedures.
10.1
Proficiency testing
10.1.1
Laboratories
shall perform proficiency testing in order to verify the laboratory's
performance. The frequency of the
proficiency testing shall be, at least, annually. Where possible, at least one of these
proficiency tests should be from a recognized external proficiency test
provider.
10.1.2
Proficiency
test samples should be representative of the laboratory's normal casework.
10.1.3
The
analytical scheme applied to the proficiency test should be in concert with
normal laboratory analysis procedures.
10.2
Competency testing
10.2.1
Laboratories
shall monitor the competency of their analysts annually.
10.2.2
If
competency test samples are utilized, they should be representative of the
laboratory's normal casework.
10.2.3
The
analytical scheme applied to the competency test should be in concert with
normal laboratory analysis procedures.
11.1
Method
validation is required to demonstrate that methods are suitable for their
intended purpose (see PART IV B – Validation).
12.1
Audits
of laboratory operations should be conducted at least once a year.
12.2
Records
of each audit shall be maintained and should include the scope, date of the
audit, name of auditor(s), findings and any necessary corrective actions.
13 Deficiency
of analysis
In the course of examining seized drug
samples and related materials, laboratories may encounter some operations or
results that are deficient in some manner.
Each laboratory shall have a documented policy to address such
deficiencies.
13.1
This
policy shall include the following:
a) a definition of a deficiency as any erroneous analytical
result or interpretation, or any unapproved deviation from an established
policy or procedure in an analysis;
NOTE Deviations from
established policy shall have documented management approval.
b) a
requirement for immediate cessation of the activity or work of the individual
involved, if warranted by the seriousness of the deficiency, as defined in the
documented policy;
c) a requirement for administrative review of the activity or
work of the individual involved;
d) a requirement for evaluation of the impact the deficiency
may have had on other activities of the individual or other analysts;
e) a requirement for documentation of the follow-up action
taken as a result of the review;
f) a requirement for communication to appropriate employees of
any confirmed deficiency which may have implications for their work.
NOTE It should be
recognized that to be effective, the definition for "deficiency of
analysis" shall be relatively broad.
As such, deficiencies may have markedly different degrees of
seriousness. For example, a
misidentification of a controlled substance would be very serious and perhaps
require that either the methodology or the analyst be suspended pending
appropriate remedial action, as determined by management. However, other deficiencies might be more
clerical in nature, requiring a simple correction at the first line supervisory
level, without any suspension of methodology or personnel. Thus, it may well be advantageous to identify
the differing levels of seriousness for deficiencies and make the action
required be commensurate with the seriousness.
14 Health and safety
Laboratories
shall have a documented health and safety program in place.
14.1
Health and safety requirements
14.1.1
All
personnel should receive appropriate health and safety training.
14.1.2
Laboratories
shall operate in accordance with laboratory policy and comply with any relevant
regulations.
14.1.3
Laboratory
health and safety manual(s) shall be readily available to all laboratory
personnel.
14.1.4
Material
Safety Data Sheets shall be readily available to all laboratory personnel.
14.1.5
All
chemicals, biohazards and supplies shall be stored and disposed of according to
applicable government regulations and laboratory policy.
14.1.6
Safety
hazards such as syringes, items with sharp edges or noxious substances should
be so labeled.
15 Additional
documentation
In
addition to casework documentation, laboratories shall maintain documentation
on the following topics:
·
test
methods/procedures for drug analysis
·
reference
materials (including source and verification)
·
preparation
and testing of reagents
·
evidence
handling protocols
·
instrument
and equipment calibration and maintenance
·
instrument
and equipment inventory (e.g., manufacturer, model, serial number, acquisition
date)
·
proficiency
testing
·
personnel
training and qualifications
·
quality
assurance protocols and audits
·
health,
safety and security protocols
·
validation
data and results
·
uncertainty data.
PART IV B
QUALITY
ASSURANCE/VALIDATION OF ANALYTICAL METHODS
1
Introduction
1.1
Definition and purpose of validation
Validation is the confirmation by examination and
the provision of objective evidence that the particular requirements for a
specific intended use are fulfilled.
There are numerous documents that address the topic of validation but
there are few validation protocols for methods specific to seized drug
analysis.
1.2
Analytical scheme
An
analytical scheme shall be comprised of validated methods that are appropriate
for the analyte.
1.2.1 The combinations of methods chosen
for a particular analytical scheme shall identify the specific drug of
interest, preclude a false positive and minimize false negatives.
1.2.2 For quantification the method
should reliably determine the amount of analyte
present.
1.2.3 If validated methods are used from published
literature or another laboratory’s protocols, then the methods shall be
verified within each laboratory.
1.2.4 If
non-routine validated methods are used, then the method shall be verified prior
to use.
1.2.5 Verification should, at a
minimum, demonstrate that a representative set of reference materials has been
carried through the process and yielded the expected results.
1.3
Individual laboratory responsibility
Each laboratory should determine whether their
current standard operating procedures have been validated, verified or require
further validation/verification.
1.4
Operational environment
All methods shall be validated or
verified to demonstrate that they will perform in the normal operational
environment when used by individuals expected to utilize the methods on
casework.
1.5
Documentation
The entire validation/verification process shall be
documented and the documentation shall be retained. Documentation shall include, but is not
limited to the following:
·
personnel involved
·
dates
·
observations from the process
·
analytical data
·
a statement of conclusions and/or recommendations
·
authorization approval signature.
1.6
Recommendation
To meet the above requirements, SWGDRUG recommends
that laboratories follow the applicable provisions of Section 2 [General
Validation Plan] when validating seized drug analytical methods.
For further information, see Supplemental Document SD-2
(Preparing Validation Plans, Section I: Analytical Techniques – Elements to
Consider and Section II: Example Validation Plan for GC/MS Identification and Quantitation of Heroin).
2
General validation plan
2.1
Purpose/scope
This is an introductory statement that
will specify what is being tested, the purpose of the testing and the result(s)
required for acceptance.
2.1.1 Performance
specification
A list of specific objectives (e.g.,
trueness and precision) should be determined prior to the validation process.
2.1.2 Process
review
After completion of the validation
process the objectives should be revisited to ensure that they have been
satisfactorily met.
2.2
Analytical method
State exactly the method to be
validated. It is essential that each
step in the method be demonstrated to perform satisfactorily. Steps that constitute a method for the
identification and/or quantification of seized drugs may include:
·
visual characterization (e.g., macroscopic
examination)
·
determination of quantity of sample, which may
include:
o
weight
o
volume
o
item count
·
sampling (representative or random, dry, homogenized, etc.)
·
stability of analyte
·
sample preparation
o
extraction method
o
dissolution
o
derivatization
o
crystallization
o
techniques for introducing sample into
instrumentation
·
instrumental parameters and specifications
o
list the instruments and equipment (e.g., balance
and glassware) utilized
o
instrument conditions
·
software applications (e.g., software
version, macros)
·
calculations
o
equation(s) to be used
o
unit specification
o
number of measurements required
o
reference values
o
significant figure conventions
o
conditions for data rejection
o
uncertainty determination.
2.3
Reference materials
Appropriate
reference material(s) shall be used for qualitative and quantitative
procedures. Traceability of the reference
material is required.
2.4
Performance
characteristics
2.4.1 Selectivity
Assess the capability of the method to
identify/quantify the analyte(s) of interest, whether
pure or in a mixture.
2.4.2
Matrix
effects
Assess the
impact of any interfering components and demonstrate that the method works in
the presence of substances that are commonly encountered in seized drug samples
(e.g. cutting agents, impurities, by-products, precursors).
2.4.3
Recovery
May be determined for quantitative
analysis.
2.4.4
Accuracy
2.4.4.1
Precision
(Repeatability/Reproducibility)
Determine
the repeatability and reproducibility of all routine methods. Conditions under which these determinations
are made shall be specified.
NOTE Reproducibility
determination may be limited to studies within the same laboratory.
2.4.4.1.1
Within the scope of the
validation, determine acceptable limits for repeatability and
reproducibility.
2.4.4.1.2 For
qualitative analysis, run the qualitative method a minimum of ten times.
2.4.4.1.3 For
quantitative analysis run the quantitative method a minimum of ten times.
2.4.4.1.4 Validation
criteria for non-routine methods may differ from what is stated above.
2.4.4.2
Trueness
Trueness shall be determined for
quantitative methods to assess systematic error. Trueness can be assessed
through various methods such as:
·
comparison of a method-generated value for the
reference material with its known value using replicate measurements at
different concentrations
·
performance of a standard addition method
·
comparison to proficiency test results
·
comparison with a different validated
analytical method.
2.4.5
Range
Determine
the concentration or sample amount limits for which the method is applicable.
2.4.5.1
Limit
of detection (LOD)
Limit of detection shall be determined
for all qualitative methods.
2.4.5.1.1 Determine the lowest amount of analyte that will be detected and can be identified.
2.4.5.1.2 The results obtained at the LOD are not
necessarily quantitatively accurate.
2.4.5.2
Limit
of quantitation (LOQ)
Limit of Quantitation
shall be determined for all quantitative methods. Determine the lowest concentration that has
an acceptable level of uncertainty.
2.4.5.3
Linearity
Linearity
shall be determined for all quantitative methods.
2.4.5.3.1 Determine the mathematical relationship
(calibration curve) that exists between concentration and response over a
selected range of concentrations.
2.4.5.3.2 The LOQ effectively forms the lower end
of the working range.
2.4.5.3.3 Determine
the level of acceptable variation from the calibration curve at various
concentrations.
2.4.5.3.4 Determine the upper limits of the
working range.
2.4.6
Robustness
Robustness shall be determined for
either qualitative or quantitative methods.
Alter method parameters individually and determine any changes to
accuracy.
2.4.7
Ruggedness
Ruggedness may be determined for either qualitative
or quantitative methods. Ruggedness
should assess the factors external to the method.
2.4.8 Uncertainty
The contribution of random and systematic errors to
method result uncertainty shall be assessed and the expanded uncertainty
derived for quantitative methods (see PART IV C –
Uncertainty).
3 Quality
control
Acceptance criteria for quality
control parameters should be adopted prior to implementation of the method.
4 References
a)
The Fitness for Purpose of
Analytical Methods, A Laboratory Guide to Method Validation and Related Topics, EURACHEM
Guide, 1998.
b)
Federal
Register, Part VIII, Department of Health and Human Services, March
1995, pages 11259-62.
c) “Validating
Analytical Chemistry Methods”, Enigma Analytical Training Course (Version
2000-1), Breckenridge, CO, 2000, pages 8-4, 8-5.
d) “Guidelines
for Forensic Science Laboratories”, ILAC-G19:2002, page 10.
PART IV C
Quality
Assurance/Uncertainty
1
Introduction
This
recommendation provides guidance on the concept of uncertainty and its
application to the qualitative and quantitative analysis of seized drugs. In this context, uncertainty encompasses
limitations of qualitative methods as well as numerical ranges as applied to
quantitative analyses.
1.1
SWGDRUG
considers an understanding of uncertainty to be fundamental to the
interpretation and reporting of results.
1.2
The
term “uncertainty” does not imply doubt; rather, its consideration provides
assurance that results and conclusions from methods and analytical schemes are
fit for purpose.
1.3
SWGDRUG
recommends the concept of uncertainty be considered for all analytical
results.
1.4
Laboratory
management shall ensure that uncertainty be addressed through the provision of
training, procedures and documentation.
1.5
Laboratory
management should consider customer requirements which influence the
application of uncertainty.
1.6
Benefits
The
benefits of determining and understanding uncertainty include:
·
Enhancing
confidence through increased understanding of results
·
Providing
a mechanism to express the reliability of results
·
Enabling
the laboratory and customer to evaluate the fitness for purpose of results
·
Facilitating
the identification of procedural limitations and providing a basis for
improvement
·
Complying
with accreditation requirements.
1.7
Application of uncertainty
Qualitative and quantitative analyses
require different approaches. Analysts shall understand the limitations of
qualitative and quantitative determinations and have tools to estimate a value
for measurement uncertainty of relevant, but not necessarily all, numerical
results. In this regard, efforts should
be made to use the vocabulary, symbols, and formatting expressed in documents
published by a Standards Developing Organization (SDO) such as ISO and ASTM International.
2
Qualitative Analysis
The
identification of seized drugs requires the combination of methods to form an
analytical scheme (see PART III B - Methods of Analysis/Drug
Identification).
2.1
Individual
methods have limitations and, consequently, uncertainty. Uncertainty of qualitative methods is not
typically expressed in numerical terms.
2.2
Understanding
these limitations enables the laboratory or analyst to build an appropriate
analytical scheme to correctly identify a drug or chemical.
2.2.1
It
is expected that, in the absence of unforeseen circumstances, an appropriate
analytical scheme effectively results in no uncertainty in reported
identifications.
2.2.2
Relevant
limitations of an analytical scheme (e.g., inability to differentiate isomers,
unavailability of reference material) should be documented and may need to be
included in the report (see Section 5.2.1).
3
Quantitative Measurements
3.1
Quantitative
measurements have an associated uncertainty, which is defined as a parameter
that “characterizes the dispersion of the values that could
reasonably be attributed to the particular quantity subject to measurement
or characteristic subject to test”
(see Glossary).
3.2
A
rigorous calculation of measurement uncertainty is not always required.
3.2.1
A
laboratory shall understand the contributing factors of measurement uncertainty
for each analytical procedure and evaluate them with respect to customer,
accreditation or jurisdictional requirements.
3.2.2
Where
a value is critical, such as a weight or purity level close to a statutory
threshold, an appropriate measurement uncertainty estimation
shall be applied.
3.3
Primary
numerical values reported in the analysis of seized drugs are weight and
purity. Where other values are measured
(e.g., size, volume, estimated tablet numbers), the same principles stated
herein apply.
4
Estimation of measurement uncertainty
for quantitative determinations
4.1
Sources of uncertainty for weight
determination
4.1.1
The
uncertainty of a reported value is dependant on the
weighing process. Factors for
consideration include:
·
Single
versus multiple items (number of weighing operations)
·
Taring
of a weighing vessel as a separate weighing operation
·
Extrapolation
of population weight from limited sampling of multiple items
·
Aggregate
weighings
·
Incomplete
recovery of material from the packaging
·
Balance
selection (e.g., readability, capacity, calibration uncertainty)
·
Balance
operation (e.g., sample placement on pan, environmental conditions).
4.1.2
For
further information and examples of estimation of measurement uncertainty for
weight determinations, see Supplemental Document SD-3 (Measurement Uncertainty for Weight
Determinations in Seized Drug Analysis).
4.2
Sources of uncertainty for purity
determination
The
uncertainty of a reported purity value is dependant
upon the entire quantitation process. Factors for consideration include:
·
Sampling
plan (e.g., handling of multiple exhibits)
o Sample homogeneity
·
Analytical
method
o Sample preparation (e.g., sample size,
matrix effects, solubility)
o Analytical technique
o Reference material (e.g., purity of
standard)
o Equipment and instrument properties
(e.g., glassware, pipetters, balances,
chromatographs)
o Concentration of analyte
o Environmental conditions.
4.3
Factors relevant to estimation of
measurement uncertainty
4.3.1
When
estimating measurement uncertainty, the following sources of error shall be considered:
4.3.1.1
Analytical
Error: Systematic and random error both
contribute to measurement uncertainty and shall be addressed through method
validation and quality assurance practices (Part IV B). SWGDRUG recommends that for all validated
procedures, systematic error is characterized and minimized.
4.3.1.2
Sampling
Error: The sample and sampling procedure
are often the greatest contributors to measurement uncertainty.
4.3.2
Where
appropriate, confidence levels (e.g., 95% or 99.7%) shall be selected based on
considerations relevant to the analytical context.
4.3.3
Uncertainty
information shall be recorded in validation documents and/or case records.
4.4
Approaches
for estimating measurement uncertainty
4.4.2
Uncertainty budget approach
4.4.2.1
In this approach all sources of error
are separately identified and tabulated.
4.4.2.2
A value is assigned to each source of
error (collectively or individually) using either:
·
empirical data (e.g., from validation
process, historical performance data, control chart data, proficiency tests)
·
published data (e.g., volumetric
glassware tolerances)
·
combination
of empirical and published data.
NOTE:
Control chart data, including measurement quality assurance, should be derived
from multiple data points over time and is expected to capture the typical
variations of realistic laboratory processes.
4.4.2.3
Where a source has an uncertainty which
is insignificant compared to other sources, it can be excluded.
4.4.2.4
The remaining significant values are
used to calculate the combined standard uncertainty and expanded uncertainty.
4.4.3
Non-budget approaches
4.4.3.1
The sources of uncertainty that are
separately assessed in the budget method are collectively assessed by
experimental measurement. In this
approach data obtained
from a statistically significant number of replicate analyses utilizing a
validated method with an appropriate sampling plan
may be utilized to calculate the standard or expanded uncertainty.
4.4.3.2
An alternate approach involves the use
of two standard deviations (2s)
of the test method results from reproducibility data from the validation
studies. This provides an approximation
of the measurement uncertainty for non-critical values.
5
Reporting
of uncertainty
5.1
Reporting
Uncertainty should be reported when it
may impact the use of a result by the customer.
Factors which influence the decision to report uncertainty include:
5.1.1
Jurisdictional
·
Prevailing statutory requirement
·
Relevant governing body (agency)
requirements
·
Customer requests
·
Potential exculpatory value
5.1.2
Types of Analysis
·
Qualitative: Qualitative results where limitations of
analytical scheme are known and relevant (e.g., inability to differentiate isomers, unavailability of
reference material)
·
Quantitative: Quantitative measurements where a value is
critical (e.g., weight or
purity level close to a statutory threshold)
5.1.3
Laboratory accreditation requirements
5.2
Reporting
Examples
Reporting requirements and styles
differ among agencies. The examples
listed below are drawn from laboratories with varied requirements.
5.2.1
Qualitative Results
5.2.1.1
Contains ephedrine or
pseudoephedrine. Item tested: 5.2 grams
net.
5.2.1.2
Visual
examination determined that the physical characteristics are consistent with a
Schedule IV pharmaceutical preparation containing Diazepam. There was no apparent tampering of the dosage
units and no further tests are being conducted.
5.2.1.3
Contains
cocaine (salt form not determined)
5.2.2
Quantitative Results
Factors to be considered when reporting
measurement uncertainty include use of significant figures, confidence
intervals and rounding/truncating of results.
5.2.2.1
Active drug ingredient (established or
common name) methamphetamine hydrochloride
Gross
weight: 25.6 grams
Net
weight: 5.2 grams
Conc.
or purity: 54.7% (±
2.8%)*
Amount
of actual drug: 2.8 grams
Reserve
weight: 5.1 grams
* This value represents the
quantitative uncertainty measurement estimate for the laboratory system.
5.2.2.2
Positive for cocaine in the sample tested
Net
weight of total sample: 5.23 grams ±
0.03 grams
Quantitation: 54.7% ± 2.8%
5.2.2.3
Sample tested positive for cocaine
Net
weight: 5.23 grams
Purity:
54.7%
Confidence
Range: ±
2.8%*
Calculated
net weight of drug: 2.8 grams of cocaine
*Confidence
range refers to a 95% confidence level.
5.2.2.4
Cocaine was identified in the Item 1
powder at a purity of 65 ±
9% (99.7% confidence level). The Item 1
powder weighed 800 ±
4 mg (99.7% confidence level).
5.2.2.5
White powder: 5.6 grams
The
range of heroin concentration identified in the sample was not less than 53.2%
and not more than 56.2%.
6
Training
6.1
Individuals responsible for
determining, evaluating and documenting uncertainty in the context of
seized-drug analysis shall be capable of competently demonstrating familiarity
with foundational concepts and principles of estimating uncertainty.
6.1.1
Useful topics to review include:
·
General metrology to include:
terminology, symbols, formulae, publications, international organizations, and
global application as related to seized-drug analysis
·
The concepts of random and systematic
error, accuracy, precision (repeatability, reproducibility, and their
conditions), statistical control, standard and expanded uncertainty, correlation
and propagation of error
·
Reporting conventions including use of
significant figures, truncation and rounding
·
Basic statistics (descriptive and
inferential) to include: measures of central tendency (e.g., median), measures
of variation, statistical modeling, sampling, probability, confidence interval,
and significance level
6.2
All analysts shall be capable of
explaining their laboratory’s procedures for evaluating uncertainty of
qualitative and quantitative analyses.
7
References
7.1
Eurachem/CITAC
Guide: The Expression of Uncertainty in
Qualitative Testing, Committee Draft September 2003.
7.2
GUM,
Evaluation of measurement data — Guide
to the expression of uncertainty in measurement Published by the Joint
Committee for Guides in Metrology (JCGM), JCGM 100:2008.
7.3
Guidelines for Evaluation and Expressing the Uncertainty of NIST
Measurement Results, National Institute of Standards and
Technology, NIST Technical Note 1297, 1994 Edition.
7.4
General requirements for the competence of testing and calibration
laboratories
International
Organization for Standardization, ISO/IEC 17025: 2005.
7.5
Guide for the use
of the International System of Units (SI), Taylor, B.N.,
National Institute of Standards and Technology, April 1995.
7.6
Standard
Practice of Using Significant Digits in Test Data to Determine Conformance with
Specifications, ASTM E29, West Conshohosken,
PA.
7.7
Quantifying
Uncertainty in Analytical Measurements, Eurachem, 2000, 2nd ED.
7.8
Experimental
Statistics, M. Natrella, National Bureau of
Standards (NBS), USA 1966.
7.9
ISO 3534-1
Statistics — Vocabulary and symbols Part
1: General statistical terms and terms used in probability, ISO 3534-2 Statistics — Vocabulary and symbols Part 2:
Applied statistics International
Organization for Standardization, Switzerland, 2006.
7.10
ISO Guide 99:2007
The International Vocabulary of Basic and
General Terms in Metrology, International Organization for Standardization,
Switzerland, 2007.
7.11
ISO 5725-1
Accuracy (Trueness and Precision) of
Measurement Methods and Results Part 1: General Principles and Definitions International Organization
for Standardization, Switzerland,1994.
7.12
The Uncertainty of Measurements.
Physical and Chemical Metrology Impact and Analysis. Kimothi, S.K., Milwaukee: American Society for Quality,
2002.
7.13
Fundamentals of Analytical Chemistry, 8th Edition, Skoog, D.A., et al. Brooks Cole, 2003.
7.14
Measurement Uncertainty Arising from Sampling: A Guide to
Methods and Approaches.
Eurachem/CITAC Guide, 1st edition,
2007.
7.15
ASTM E2655 Standard Guide for Reporting Uncertainty of Test Results and Use of the
Term Measurement Uncertainty in ASTM Test Methods.
ANNEX
A
A.1
Introduction
This
glossary of terms and definitions has been developed and adopted by the SWGDRUG
core committee from a variety of sources that are listed in endnotes. In some instances, the core committee
modified existing definitions or created definitions where none could be found
in standard references.
A.2
Terms and definitions
A.2.1
accuracy
closeness of agreement between a test result or
measurement result and the true value
NOTE 1 In practice, the accepted
reference value is substituted for the true value.
NOTE 2 The term
“accuracy”, when applied to a set of test or measurement results, involves a
combination of random components and a common systematic error or bias
component.
NOTE 3 Accuracy
refers to a combination of trueness and
precision.
[ISO 3534-2:2006]
A.2.2
analyst
a designated person
who:
·
examines and analyzes seized
drugs or related materials, or directs such examinations to be done,
·
independently has access to
unsealed evidence in order to remove samples from the evidentiary material for
examination and,
·
as a
consequence of such examinations, signs reports for court or other purposes [SWGDRUG]
A.2.3
analyte
the component of a system to be analyzed
[IUPAC]
A.2.4
audit
systematic, independent and documented process for obtaining audit evidence
and evaluating it objectively to determine the extent to which audit criteria
are fulfilled
[ISO 9000:2005 (E)]
A.2.5
bias
the difference between the expectation of the test
results and an accepted reference value.
[ASTM E
177-06b, ASTM
E456-06]
A.2.6
blank
specimen or sample not containing
the analyte or other interfering substances
[Modified UNODC Definition]
A.2.7
byproduct
a secondary or incidental product of a manufacturing process.
[Collins English Dictionary - Complete &
Unabridged 10th Edition]
A.2.8
calibration
operation that, under specified conditions, in
a first step, establishes a relation between the quantity values with measurement
uncertainties provided by measurement
standards and corresponding indications
with associated measurement
uncertainties and, in a second step, uses this information
to establish a relation for obtaining a measurement result from an indication
NOTE 1 A calibration
may be expressed by a statement, calibration function, calibration diagram, calibration curve, or calibration table. In
some cases, it may consist of an additive or multiplicative correction of the indication with
associated measurement uncertainty.
NOTE 2 Calibration
should not be confused with adjustment
of a measuring system, often mistakenly called “self-calibration”, nor with verification
of calibration.
[VIM 2008]
A.2.9
capacity
the amount of finished product that could be produced, either in one
batch or over a defined period of time, and given a set list of variables.
[SWGDRUG]
A.2.10
catalyst
a substance whose presence initiates or changes the rate of a
chemical reaction, but does not itself enter into the reaction.
[ASTM-D6161]
reference material characterized by a metrologically valid procedure for one or more specified
properties, accompanied by a certificate that provides the value of the
specified property, its associated uncertainty, and a statement of metrological
traceability
NOTE 1 The concept of value includes qualitative attributes such as
identity or sequence. Uncertainties for such attributes may be expressed as
probabilities.
NOTE 2 Metrologically valid procedures for the production and
certification of reference materials are given in, among others, ISO Guides 34
and 35.
NOTE 3 ISO Guide 31
gives guidance on the contents of certificates.
NOTE 4 VIM has an
analogous definition (ISO/IEC Guide 99:2007, 5.14).
[ISO GUIDE 30:2008]
A.2.12
chain of custody
procedures and documents that account for the integrity of a specimen or
sample by tracking its handling and storage from its point of collection to its
final disposition
[UNODC]
A.2.13
clandestine
secret and concealed, often for illicit reasons.
[Collins English Dictionary - Complete &
Unabridged]
A.2.14
combined
standard uncertainty
standard uncertainty of the result of
a measurement when that result is obtained from the values of a number of other
quantities, equal to the positive square root of a sum of terms, the terms
being the variances or covariances of these other
quantities weighted according to how the measurement result varies with changes
in these quantities
[GUM 2008]
A.2.15
control
material of established origin that is used to evaluate the performance of
a test or comparison
[ASTM E1732-09]
A.2.16
deficiency of analysis
any erroneous analytical result or interpretation, or any unapproved
deviation from an established policy or procedure in an analysis
[SWGDRUG]
A.2.17
detection
limit
the lowest concentration of analyte in a sample that can be detected, but not
necessarily quantitated under the stated conditions
of the test
[EURACHEM]
A.2.18
expanded
uncertainty (U)
quantity defining an interval about a result
of a measurement that may be expected to encompass a large fraction of the
distribution of values that could reasonably be attributed to the measurand
NOTES
1. The fraction may
be regarded as the coverage probability or level of confidence of the interval.
2. To associate a
specific level of confidence with the interval defined by the expanded
uncertainty requires explicit or implicit assumptions regarding the probability
distribution characterized by the measurement result and its combined standard
uncertainty. The level of confidence that may be attributed to this interval
can be known only to the extent to which such assumptions can be justified.
3. An expanded
uncertainty U is calculated from a combined standard uncertainty uc and coverage factor k
using: U = k x uc
[EURACHEM, GUM 2008]
[SWGDRUG]
A.2.20
false positive
test result that states that an analyte
is present, when, in fact, it is not present or, is present in an amount less
than a threshold or designated cut-off concentration
[SWGDRUG]
A.2.21
finished product
a manufactured product ready for use.
[SWGDRUG]
A.2.22
intermediate
substance that is manufactured for and consumed in or used for
chemical processing to be transformed into another substance.
[ASTM- F2725]
A.2.23
limit
of detection
see A.2.13 detection
limit
A.2.24
limit
of quantitation
the
lowest concentration of an analyte that can be
determined with acceptable precision (repeatability) and accuracy under the
stated conditions of the test
[EURACHEM]
A.2.25
linearity
defines the ability of the method to
obtain test results proportional to the concentration of analyte
NOTE The Linear Range is by inference the
range of analyte concentrations over which the method
gives test results proportional to the concentration of the analyte.
[EURACHEM]
A.2.26
pharmaceutical identifiers
physical characteristics of
tablets, capsules or packaging indicating the identity, manufacturer, or
quantity of substances present
[SWGDRUG]
A.2.27
population
the totality of items or units of
material under consideration
[ASTM
E456-06]
A.2.28
precision
closeness of agreement between independent test/measurement results obtained under stipulated conditions
NOTE 1 Precision
depends only on the distribution of random errors and does not relate to the true value or the specified
value.
NOTE 2 The measure of
precision is usually expressed in terms of imprecision and computed as a
standard deviation of the test results or
measurement results. Less precision is reflected by a
larger standard deviation.
NOTE 3 Quantitative
measures of precision depend critically on the stipulated conditions. Repeatability conditions and reproducibility conditions are particular sets
of extreme stipulated conditions.
[ISO 3534-2:2006]
A.2.29
precursor
a chemical that is transformed into another compound, as in
the course of a chemical reaction, and therefore precedes that compound in the
synthetic pathway.
[Webster’s
Unabridged Dictionary of the English Language]
A.2.30
procedure
specified way to carry out an activity or process
NOTES
1. Procedures can be documented or not.
2. When a procedure is documented, the term
“written procedure” or “documented procedure” is frequently used. The document that contains a procedure can be
called a “procedure document.”
[ISO 9000:2005 (E)]
A.2.31
proficiency testing
ongoing
process in which a series of proficiency specimens or samples, the
characteristics of which are not known to the participants, are sent to
laboratories on a regular basis. Each
laboratory is tested for its accuracy in identifying the presence (or concentration)
of the drug using its usual procedures.
An accreditation body may specify participation in a particular
proficiency testing scheme as a requirement of accreditation.
[UNODC]
A.2.32
qualitative analysis
analysis in which
substances are identified or classified on the basis of their chemical or
physical properties, such as chemical reactivity, solubility, molecular weight,
melting point, radiative properties (emission,
absorption), mass spectra, nuclear half-life, etc. See also A.2.29 quantitative analysis
[IUPAC]
A.2.33
quality assurance
part of quality management focused on providing confidence that
quality requirements will be fulfilled.
[ISO 9000:2005 (E)]
A.2.34
quality management
coordinated activities to direct and
control an organization with regard to quality
NOTE Direction and control with regard to quality
generally includes establishment of the quality policy and quality objectives,
quality planning, quality control, quality assurance and quality improvement.
[ISO 9000:2005 (E)]
A.2.35
quality
manual
document specifying the quality
management system of an organization
NOTE Quality manuals can vary in detail and format
to suit the size and complexity of an individual organization.
[ISO 9000:2005 (E)]
A.2.36
quantitative
analysis
analyses in which the amount or concentration
of an analyte may be determined (estimated) and
expressed as a numerical value in appropriate units. Qualitative
analysis may take place without quantitative analysis, but quantitative
analysis requires the identification (qualification) of the analytes
for which numerical estimates are given
[IUPAC]
A.2.37
random sample
the sample so selected that
any portion of the population has an equal (or known) chance of being
chosen. Haphazard or arbitrary choice of
units is generally insufficient to guarantee randomness
[IUPAC]
A.2.38
reagent
a chemical used to react with another chemical, often to
confirm or deny the presence of the second chemical.
[ASTM-E1605]
A.2.39
recovery
term used in analytical and preparative chemistry to denote the
fraction of the total quantity of a substance recoverable following a chemical
procedure
[IUPAC]
A.2.40
reference
material (RM)
material, sufficiently homogeneous and stable
with respect to one or more specified properties, which has been established to
be fit for its intended use in a measurement process
NOTE 1 RM is a
generic term.
NOTE 2 Properties can
be quantitative or qualitative, e.g. identity of substances or species.
NOTE 3 Uses may
include the calibration of a measurement system, assessment of a measurement
procedure, assigning values to other materials, and quality control.
NOTE 4 A single RM
cannot be used for both calibration and validation of results in the same
measurement procedure.
NOTE 5 VIM has an
analogous definition (ISO/IEC Guide 99:2007, 5.13), but restricts the term
“measurement” to apply to quantitative values and not to qualitative
properties. However, Note 3 of ISO/IEC Guide 99:2007, 5.13, specifically
includes the concept of qualitative attributes, called “nominal properties”.
[ISO GUIDE 30:2008]
A.2.41
repeatability (of
results of measurements)
closeness of the agreement between the results
of successive measurements of the same measurand
carried out subject to all of the following conditions:
- the same
measurement procedure;
- the same
observer;
- the same
measuring instrument, used under the same conditions;
- the same
location;
- repetition
over a short period of time.
[ISO GUIDE 30:1992]
A.2.42
reproducibility (of results of measurements)
Closeness of the agreement between the
results of measurements of the same measurand, where the
measurements are carried out under changed conditions such as:
- principle
or method of measurement;
- observer;
- measuring instrument;
- location;
- conditions
of use;
- time.
[ISO GUIDE 30:1992]
A.2.43
robustness
the
robustness of an analytical procedure is a measure of its capacity to remain
unaffected by small, but deliberate variations in method parameters and
provides an indication of its reliability during normal usage
[EURACHEM]
A.2.44
ruggedness
The ruggedness of an analytical method is the degree of
reproducibility of test results obtained by the analysis of the same samples
under a variety of conditions, such as different laboratories, analysts,
instruments, lots of reagents, elapsed assay times, assay temperatures, or
days. Ruggedness is normally expressed
as the lack of influence on test results of operational and environmental
variables of the analytical method. Ruggedness
is a measure of reproducibility of test results under the variation in
conditions normally expected from laboratory to laboratory and from analyst to
analyst.
[USP
28:2005]
A.2.45
sample
subset of a population made up of one or more sampling units
NOTE 1 The sampling units could be items, numerical values or even
abstract entities depending on the population of interest.
NOTE 2 The definition of sample in ISO 3534-2 includes an example
of a sampling frame which is essential in drawing a random sample from a finite
population.
[ISO 3534-1:2006(E/F)]
act of drawing or constituting a sample
[ISO 3534-2:2006]
A.2.47
sampling plan
a specific plan which
states the sample size(s) to be used and the associated criteria for accepting
the lot
NOTES
1. A criterion is, for
example, that the number of nonconforming items is less than or equal to the
acceptance number.
2. The sampling plan does
not contain the rules on how to take the sample.
[ISO 3534-2:1993 (E/F)]
A.2.48
sampling procedure
operational requirements and/or
instructions relating to the use of a particular sampling plan; i.e., the
planned method of selection, withdrawal and preparation of sample(s) from a lot
to yield knowledge of the characteristic(s) of the lot
[ISO 3534-2:1993 (E/F)]
A.2.49
sampling scheme
a combination of sampling
plans with rules for changing from one plan to another
NOTE Some schemes have switching rules for
automatic change to tightened inspection plans or reduced inspection plans or
change to 100 % inspection.
[ISO 3534-2:1993 (E/F)]
A.2.50
selectivity (in analysis)
1. (Qualitative): The extent to which other
substances interfere with the
determination of a substance according
to a given procedure.
2. (Quantitative): A term used in conjunction
with another substantive (e.g.
constant, coefficient, index,
factor, number) for the quantitative characterization of interferences.
[IUPAC]
A.2.51
standard
uncertainty
uncertainty of the result of a measurement
expressed as a standard deviation
[GUM 2008]
A.2.53
trueness
closeness of agreement between the expectation
of a test result or a measurement result
and a true value
NOTE 1 The measure of trueness is usually expressed in terms of bias.
NOTE 2 Trueness is
sometimes referred to as “accuracy of the mean”. This usage is not recommended.
NOTE 3 In practice, the accepted reference value is substituted for
the true value.
[ISO 3534-2:2006]
A.2.54
uncertainty (measurement)
parameter, associated with the measurement result, or test result, that characterizes the dispersion
of the values that could reasonably be attributed to the particular quantity
subject to measurement or characteristic subject
to test
NOTE 1 This
definition is consistent with VIM but differs from it in phrasing to fit into
this part of ISO 3534 concepts and to include the testing of characteristics.
NOTE 2 “Parameter” is
defined in ISO 3534-1. The parameter can be, for example, a standard deviation
or a given multiple of it.
NOTE 3 Uncertainty of
measurement or test comprises, in general, many components. Some of these
components can be estimated on the basis of the statistical distribution of the
results of a series of measurements and can be characterized by standard
deviations. Other components, which can also be characterized by standard deviations,
are evaluated from assumed probability distributions based on experience or
other information.
NOTE 4 Components of
uncertainty include those arising from systematic effects associated with
corrections and reference standards which contribute to the dispersion.
NOTE 5 Uncertainty is
distinguished from an estimate attached to a test or measurement result that
characterizes the range of values within which the expectation is asserted to
lie. The latter estimate is a measure of precision rather than of accuracy and should be used only when the true value is not defined. When
the expectation is used instead of the true value, the expression “random
component of uncertainty” is used.
[ISO 3534-2:2006]
A.2.55
uncorrelated techniques
Uncorrelated
techniques are those that yield uncorrelated measurements. In practice this is often achieved by using
techniques that have a different fundamental mechanism for characterization. For example, a gas chromatographic test based
on a partition mechanism and a thin layer chromatographic system based on an
adsorption mechanism would be considered uncorrelated techniques, but two gas
chromatographic tests based on a partition mechanism would not.
[SWGDRUG]
A.2.56
validation
confirmation,
through the provision of objective evidence, that the requirements for a
specific intended use or application have been fulfilled
NOTES
1. The term “validated” is
used to designate the corresponding status.
2. The use conditions for validation
can be real or simulated.
[ISO 9000:2005(E)]
A.2.57
verification
confirmation,
through the provision of objective evidence, that specified requirements have been
fulfilled
NOTES
1. The term “verified” is
used to designate the corresponding status.
2. Confirmation can
comprise activities such as
·
performing alternative calculations,
·
comparing a new design specification with a similar proven design
specification,
·
undertaking tests and demonstrations, and
·
reviewing
documents prior to issue.
[ISO 9000:2005(E)]
A.2.58
yield, expected
the quantity of material or the percentage of theoretical yield
anticipated at any appropriate phase of production based on previous
laboratory, pilot scale, or manufacturing data.
[ASTM-E2363]
A.2.59
yield,
theoretical
the quantity that would be produced at any appropriate phase of
production based upon the quantity of material to be used, in the absence of
any loss or error in actual production.
[ASTM-E2363]
A.3
Bibliography
A.3.1
ASTM D6161-10 Standard
Terminology Used for Microfiltration, Ultrafiltration,
Nanofiltration and Reverse Osmosis Membrane Processes,
© ASTM International (ASTM), 100 Barr Harbor Drive, West
Conshohocken, PA 19428.
A.3.2
ASTM
E456-08 Standard Terminology Relating to Quality and Statistics
Annual Book of ASTM Standards 2005, Section Fourteen, General Methods and
Instrumentation, Vol 14.02 Published by the American
Society For Testing And Materials International USA.
A.3.3
ASTM E1605-04 Standard Terminology Relating to Lead in
Buildings, © ASTM International (ASTM), 100 Barr Harbor Drive, West
Conshohocken, PA 19428.
A.3.4
ASTM E1732-09 Standard Terminology Relating to
Forensic Science Annual Book of ASTM Standards 2005, Section
Fourteen, General Methods and Instrumentation, Vol
14.02 Published by the American Society For Testing And Materials International
USA.
A.3.5
ASTM E2363-06A Standard Terminology Relating to Process
Analytical Technology in the Pharmaceutical Industry, © ASTM International
(ASTM), 100 Barr Harbor Drive, West Conshohocken, PA 19428.
A.3.6
ASTM F2725-11 Standard Guide for European Union's Registration, Evaluation, and
Authorization of Chemicals (REACH) Supply Chain Information Exchange, ©
ASTM International (ASTM), 100 Barr Harbor Drive, West Conshohocken,
PA 19428.
A.3.7
Collins English Dictionary –
Complete and Unabridged, 10th Ed., New York, New York, HarperCollins
Publishers, 2003.
A.3.8
EURACHEM,
The Fitness for Purpose of Analytical
Methods English Edition, Developed by EURACHEM Working Group. Supported in part under contract with UK
Department of Trade and Industry as part of the National Measurement System
Valid Analytical Measurement (VAM) Programme 1998.
A.3.9
ISO 3534-1:2006 (E/F), INTERNATIONAL STANDARD 2nd
ed. Statistics
– Vocabulary and
symbols Part 1: General statistical terms and terms used in probability, Published
by the International Organization for Standardization, Switzerland 2006.
A.3.10
ISO
3534-2:2006 (E/F), INTERNATIONAL STANDARD 2nd ed.Statistics — Vocabulary and symbols Part 2: Applied statistics Published by the International Organization
for Standardization, Switzerland 2006.
A.3.11
ISO GUIDE
30:1992 (E/F), GUIDE 30 Terms and definitions used in
connection with reference materials 2nd Ed., Published by the
International Organization for Standardization, Switzerland 1992. ISO GUIDE
30:1992(E)/Amd.1:2008, Amendment 1 Revision of definitions
for reference material and certified reference material.
A.3.12
GUM, Evaluation of measurement data — Guide to the
expression of uncertainty in measurement Published by
the Joint Committee
for Guides in Metrology (JCGM), JCGM
100:2008.
A.3.13
ISO
9000:2005 (E), INTERNATIONAL STANDARD Quality
management systems—Fundamentals and vocabulary Published by the
International Organization for Standardization, Switzerland 2005.
A.3.14
IUPAC, Entries are from the
online version of the IUPAC Compendium of Chemical Terminology that mostly corresponds
to the second edition (1997), compiled by Alan D. McNaught
and Andrew Wilkinson (Royal Society of Chemistry, Cambridge, UK).
A.3.15
SWGDRUG, Definitions developed by
the core committee of the Scientific Working Group for the Analysis of Seized
Drugs.
A.3.16
UNODC,
GLOSSARY OF TERMS FOR QUALITY ASSURANCE AND GOOD LABORATORY PRACTICES
Published by The United Nations International Drug
Control Programme, Vienna 1995.
A.3.17
USP
28:2005, THE UNITED STATES PHARMACOPOEIA 28TH Rev. Published
by the United States Pharmacopoeial Convention Inc. USA
2005.
A.3.18
VIM,
International Vocabulary of Basic and General Terms in Metrology
Published by the International Organization for Standardization, Switzerland.
A.3.19
Webster’s Unabridged Dictionary of
the English Language, New York, New York, Random House Reference Press, 2001.
End of
Document
